Single Cell Pipeline¶
For installation guide see INSTALL
Introduction¶
The single cell pipeline performs analysis of single cell sequencing data producing:
- aligned bams
- somatic copy number changes
- qc metrics
- SNVs, breakpoints and haplotype allele counts
CLI¶
The above analyses are implemented as a series of subcommands. Each subcommand takes as input an inputs yaml file describing both the location of input files and the metadata for those input files. Subcommands also take as input a directory or directories in which results will be output.
Inputs¶
Inputs are provided as an input yaml file, with formats specific to each subcommand described below. Absolute paths are expected.
Results¶
Results for each subcommand follow a similar structure.
Each directory will contain .csv.gz, .pdf, .bam and other files produced for that set of results. In addition,
a metadata.yaml file at the root of each results directory will list of the files and provide metadata for the
set of results contained within that directory. The metadata.yaml file has 2 mandatory top level sections:
filenames listing the files in the results relative to the root directory, and meta containing at a minimum the
type of subcommand, the version of the single cell pipeline that produced the results, and which of the files in
filenames is the input yaml.
CSV table format¶
CSV tables are produced by many of the pipeline subcommands. Per column type information is provide for each .csv.gz
file as a .csv.gz.yaml file. The yaml file has the following format:
columns:
- dtype: str
name: column1
- dtype: int
name: column2
- dtype: bool
name: column3
- dtype: float
name: column4
header: true
sep: ','
Accepted dtype values are str, int, bool and float. Default separator is , but a single character can be provided
in the sep field. The presense of a header is indicated with the header: true or header: false.
The .csv.gz.yaml files are catalogued along with their .csv.gz files in the metadata.yaml file for results.
Conventions¶
Shahlab operations uses a jira ticket to track runs of the single cell pipeline on new data. The SC-1234 that appears
in results and input paths below is a placeholder for a jira ticket id.
Cell ids follow the format {sample_id}_{library_id}_R{row}_C{column}, however this is not required by the pipeline.
1. Alignment¶
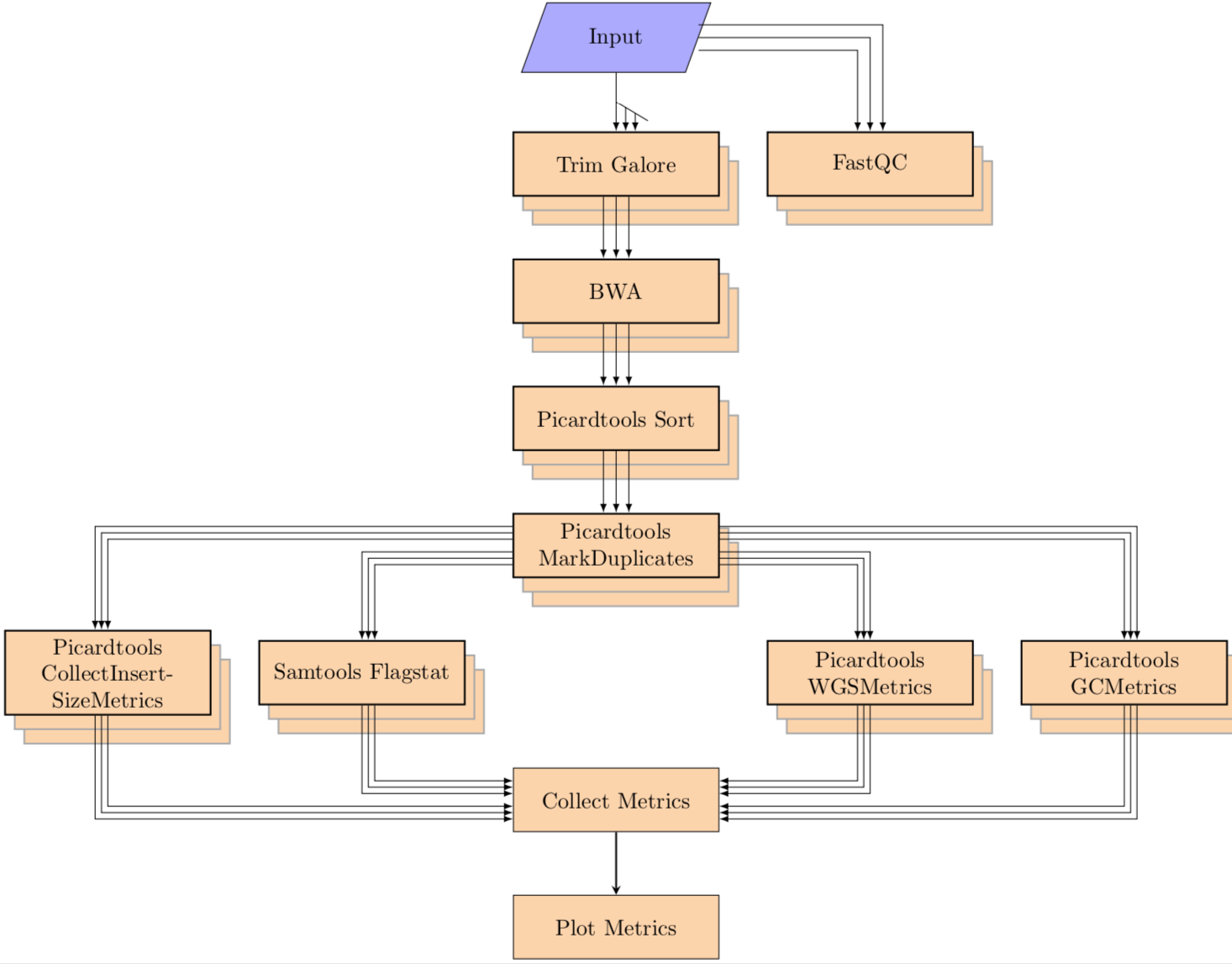 align
align
The single cell analysis pipeline runs on a list of pairs of fastq file (paired end) and performs the following steps:
Alignment:
- Run fastqc on the fastq files
- Align the fastq pairs with bwa (supports both mem and aln)
- Merge all lanes together
- create targets and realign around those targets with GATK
- sort bams files, mark duplicate reads and index final bams
- collect metrics from samtools flagstat, picardtools CollectInsertSizeMetrics, picardtools CollectWgsMetrics and picardtools CollectGcBiasMetrics
- generate QC plot
Input¶
The pipeline accepts a yaml file as input. The yaml file contains the input paths and metadata for each cell, the format for each cell is as follows:
SA12345-A12345-R01-C01:
column: 01 # column number of the well on chip
condition: A # condition during experiment. will be renamed to experimental_condition in output
fastqs:
LANE_ID_1:
fastq_1: /path/to/fastqfile/ACTACT-AGTAGT_1.fastq.gz
fastq_2: /path/to/fastqfile/ACTACT-AGTAGT_1.fastq.gz
sequencing_center: CENTERID # sequencing center id
sequencing_instrument: INSTRUMENT_TYPE # sequencing machine (HiseqX for hiseq, N550 for nextseq machine). pipeline will not run trim galore on N550 output.
LANE_ID_2:
fastq_1: /path/to/fastqfile/ATTATT-ACTACT_1.fastq.gz
fastq_2: /path/to/fastqfile/ATTATT-ACTACT_1.fastq.gz
sequencing_center: CENTERID
sequencing_instrument: INSTRUMENT_TYPE
img_col: 10 # column number of the well on chip image
index_i5: i5-INDEX
index_i7: i7-INDEX
pick_met: CELLCALL # describes whether cell is dividing, dead etc. will be renamed in output to cell_call
primer_i5: ACTACTATT
primer_i7: AGTAGTACT
row: 01 # row number of the well on chip
sample_type: C # sample type (flagged by wet lab team)
Run:¶
single_cell alignment \
--input_yaml inputs/SC-1234/fastqs.yaml \
--tmpdir temp/SC-1234/tmp \
--pipelinedir pipeline/SC-1234 \
--out_dir results/SC-1234/results/alignment \
--bams_dir results/SC-1234/results/bams \
--library_id A123123 \
...
Outputs¶
The aligment pipeline produces two sets of results, Bams and Alignment stats.
BAMs¶
Bam files are named as {CELL_ID}.bam in the BAM results directory.
The metadata file is structured as follows:
filenames:
- {CELL_ID_1}.bam
- {CELL_ID_1}.bam.bai
- {CELL_ID_2}.bam
- {CELL_ID_2}.bam.bai
...
meta:
type: cellbams
version: v0.0.0
library_id: LIBRARY_ID
cell_ids:
- CELL_ID_1
- CELL_ID_1
lane_ids:
- LANE_ID_1
- LANE_ID_2
bams:
template: {cell_id}.bam
instances:
- cell_id: CELL_ID_1
- cell_id: CELL_ID_2
Alignment Stats¶
The aligment pipeline produces a set of csv format tables and pdfs:
- alignment metrics:
{LIBRARY_ID}_alignment_metrics.csv.gz - gc metrics:
{LIBRARY_ID}_gc_metrics.csv.gz - fastqscreen metrics:
{LIBRARY_ID}_detailed_fastqscreen_metrics.csv.gz - alignment metrics plots:
{LIBRARY_ID}_alignment_metrics.pdf - alignment metrics tar:
{LIBRARY_ID}_alignment_metrics.tar.gz
The metadata file is structured as follows:
filenames:
- {LIBRARY_ID}_{TABLE_1}.csv.gz
- {LIBRARY_ID}_{TABLE_1}.csv.gz.yaml
- input.yaml
...
meta:
cell_ids:
- CELL_ID_1
- CELL_ID_2
command: 'single_cell alignment ...'
input_yaml: input.yaml
type: alignment
version: v0.0.0
library_id: LIBRARY_ID
lane_ids:
- LANE_ID_1
- LANE_ID_2
The alignment pipeline classifies the species for each read. You can read more about it in detail here: Organism Filter
You can also read more about the alignment metrics data here and gc metrics here
2. HMMcopy¶
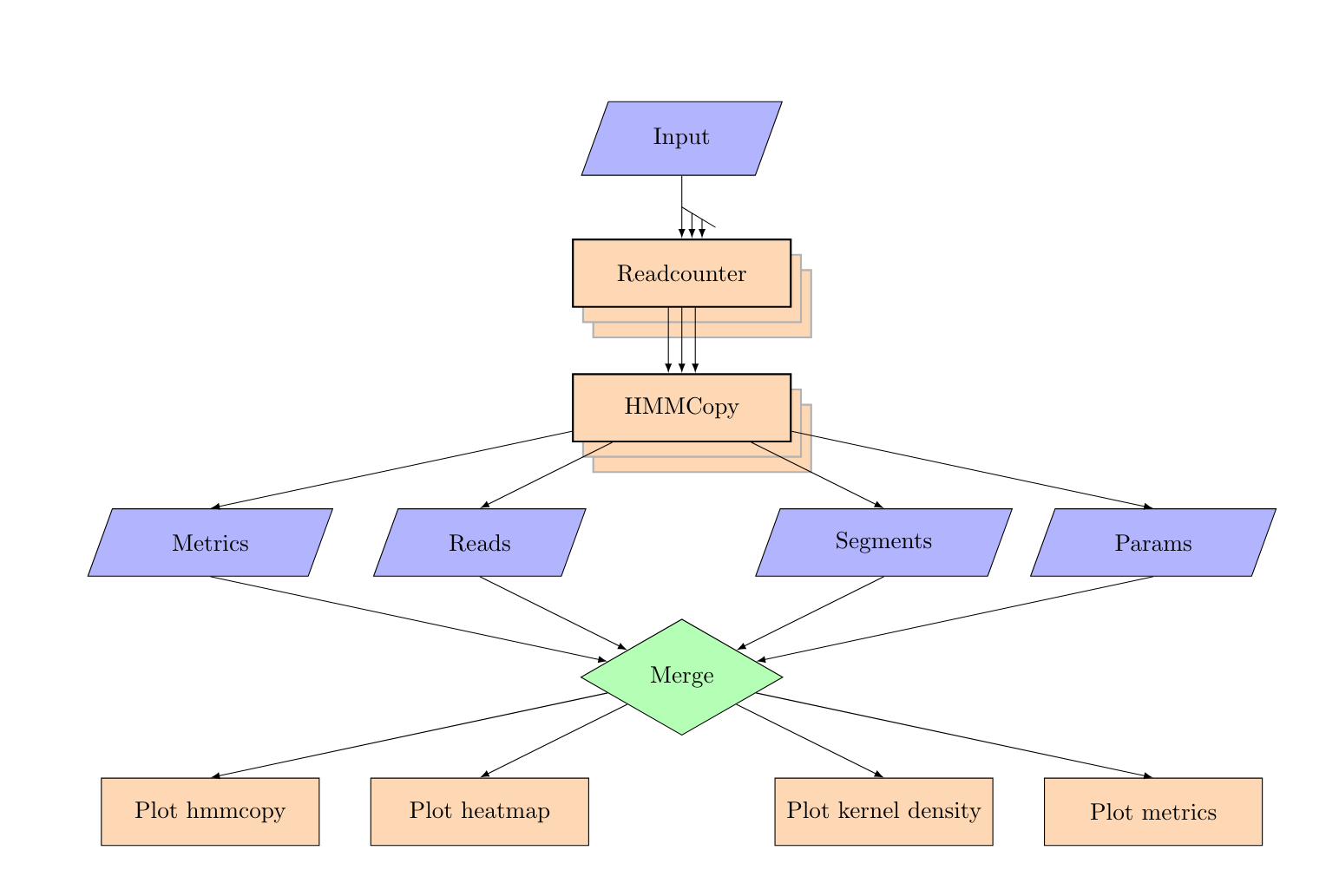 align
align
Hmmcopy:
- generate read count wig files from the bam files
- perform GC correction
- Run Hmmcopy to predict copynumber states
- generate segment and bias plots, kernel density plot and heatmaps
Input¶
The pipeline accepts a yaml file as input. The yaml file contains the input paths and metadata for each cell, the format for each cell is as follows:
SA12345-A12345-R01-C01:
bam: /path/to/aligned/SA12345-A12345-R01-C01.bam # path to the bam file, align mode will write a bam file to this path
column: 01 # column number of the well on chip
condition: A # condition during experiment. will be renamed to experimental_condition in output
img_col: 10 # column number of the well on chip image
index_i5: i5-INDEX
index_i7: i7-INDEX
pick_met: CELLCALL # describes whether cell is dividing, dead etc. will be renamed in output to cell_call
primer_i5: ACTACTATT
primer_i7: AGTAGTACT
row: 01 # row number of the well on chip
sample_type: C # sample type (flagged by wet lab team)
Run:¶
single_cell hmmcopy \
--input_yaml inputs/SC-1234/bams.yaml \
--tmpdir temp/SC-1234/tmp \
--pipelinedir pipeline/SC-1234 \
--out_dir results/SC-1234/results/hmmcopy \
--library_id A123123 \
...
Outputs¶
The hmmcopy pipeline produces HMMCopy results.
HMMCopy results¶
The output of hmmcopy is a set of tables and plots:
- reads table:
{LIBRARY_ID}_reads.csv.gz - segs table:
{LIBRARY_ID}_segments.csv.gz - params table:
{LIBRARY_ID}_params.csv.gz - metrics table:
{LIBRARY_ID}_hmmcopy_metrics.csv.gz - hmmcopy data tar:
{LIBRARY_ID}_hmmcopy_data.tar.gz - igv table:
{LIBRARY_ID}_igv_segments.seg - segs plots:
{LIBRARY_ID}_segs.tar.gz - bias plots:
{LIBRARY_ID}_bias.tar.gz - heatmap plots:
{LIBRARY_ID}_heatmap_by_ec.pdf - metrics plots:
{LIBRARY_ID}_hmmcopy_metrics.pdf - kernel density plots:
{LIBRARY_ID}_kernel_density.pdf
The metadata file is structured as follows:
filenames:
- {LIBRARY_ID}_{TABLE_1}.csv.gz
- {LIBRARY_ID}_{TABLE_1}.csv.gz.yaml
...
meta:
command: 'single_cell hmmcopy ...'
input_yaml: input.yaml
type: hmmcopy
version: v0.0.0
library_id: LIBRARY_ID
cell_ids:
- CELL_ID_1
- CELL_ID_1
You can read more about the data in the reads table here, segments table here and metrics table here here.
3. Annotation¶
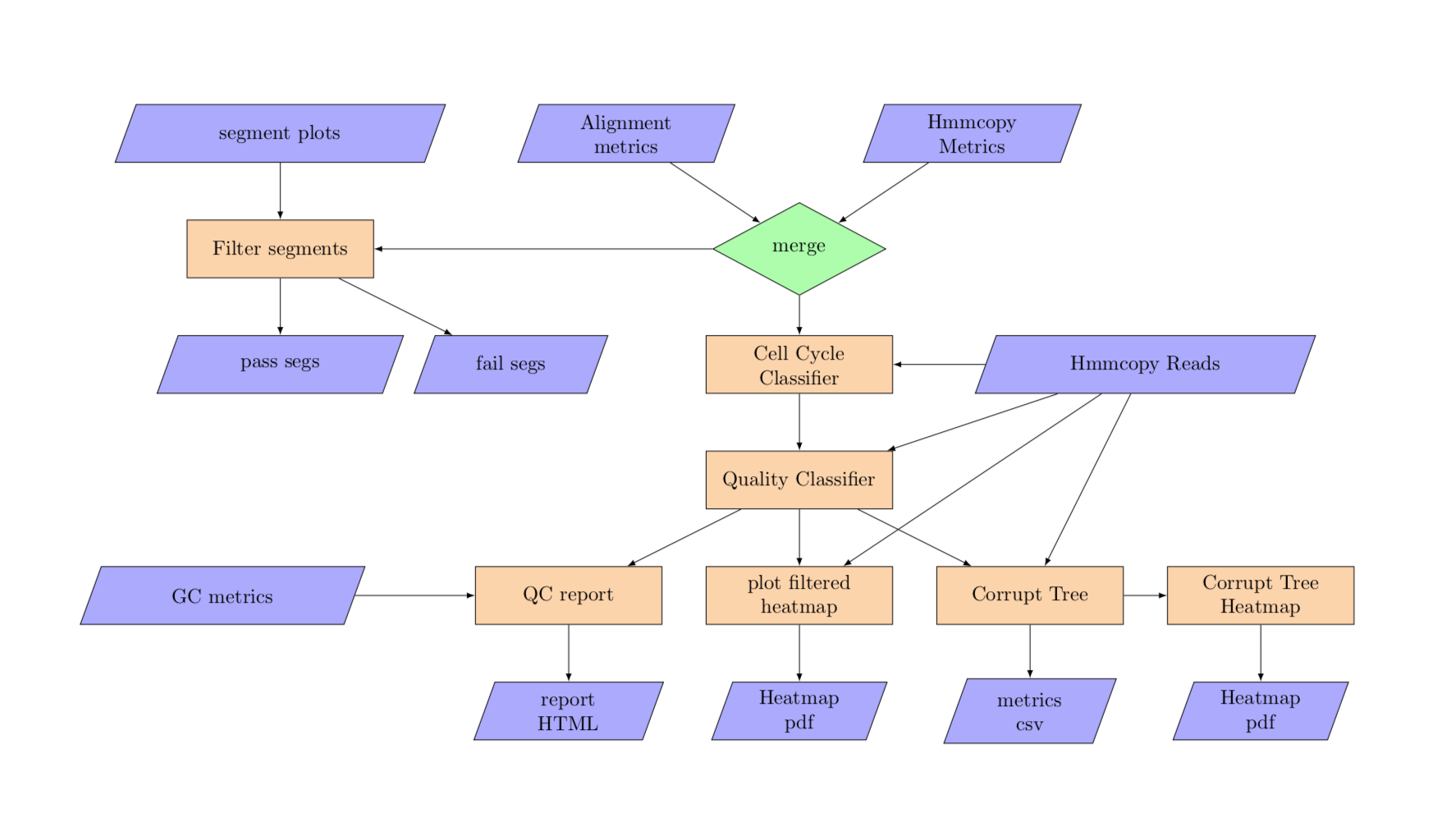 annotation
annotation
Annotation:
- Assign a quality score to each cell
- Assign cell state to each cell
- Generate a consolidated table with all metrics frim hmmcopy, alignment and from annotation
Input¶
The pipeline accepts a yaml file as input. The yaml file contains the input paths and metadata for each cell, the format for each cell is as follows:
hmmcopy_metrics: results/hmmcopy/A12345A_hmmcopy_metrics.csv.gz
hmmcopy_reads: results/hmmcopy/A12345_reads.csv.gz
alignment_metrics: results/alignment/A12345_alignment_metrics.csv.gz
gc_metrics: results/alignment/A12345_gc_metrics.csv.gz
segs_pdf_tar: results/hmmcopy/A12345_segs.tar.gz```
Run¶
single_cell annotation \
--input_yaml inputs/SC-1234/annotation.yaml \
--tmpdir temp/SC-1234/tmp \
--pipelinedir pipeline/SC-1234 \
--out_dir results/SC-1234/results/annotation \
--library_id A123123 \
...
Outputs¶
The annotation pipeline produces a set of annotation results.
Annotation results¶
The output of annotation is a set of tables and plots:
- metrics table:
{LIBRARY_ID}_metrics.csv.gz - qc report:
{LIBRARY_ID}_QC_report.html - corrupt tree newick:
{LIBRARY_ID}_corrupt_tree.newick - consensus tree newick:
{LIBRARY_ID}_corrupt_tree_consensus.newick - phylo table:
{LIBRARY_ID}_phylo.csv - loci rank trees:
{LIBRARY_ID}_rank_loci_trees.csv - filtered data table:
{LIBRARY_ID}_filtered_data.csv - corrupt tree plot:
{LIBRARY_ID}_corrupt_tree.pdf - segs pass plots:
{LIBRARY_ID}_segs_pass.tar.gz - segs fail plots:
{LIBRARY_ID}_segs_fail.tar.gz - corrupt heatmat plot:
{LIBRARY_ID}_heatmap_corrupt_tree.pdf - heatmap filt plot:
{LIBRARY_ID}_heatmap_by_ec_filtered.pdf
The metadata file is structured as follows:
filenames:
- {LIBRARY_ID}_{TABLE_1}.csv.gz
- {LIBRARY_ID}_{TABLE_1}.csv.gz.yaml
...
meta:
command: 'single_cell annotation ...'
input_yaml: input.yaml
type: annotation
version: v0.0.0
library_id: LIBRARY_ID
The annotation pipeline runs the:
- Cell Cycle Classifier: You can learn more here: cell_cycle_classifier
- Cell Quality Classifier: You can learn more here: cell quality classifier
The metrics table is described in detail here
4. Split merge cell bams¶
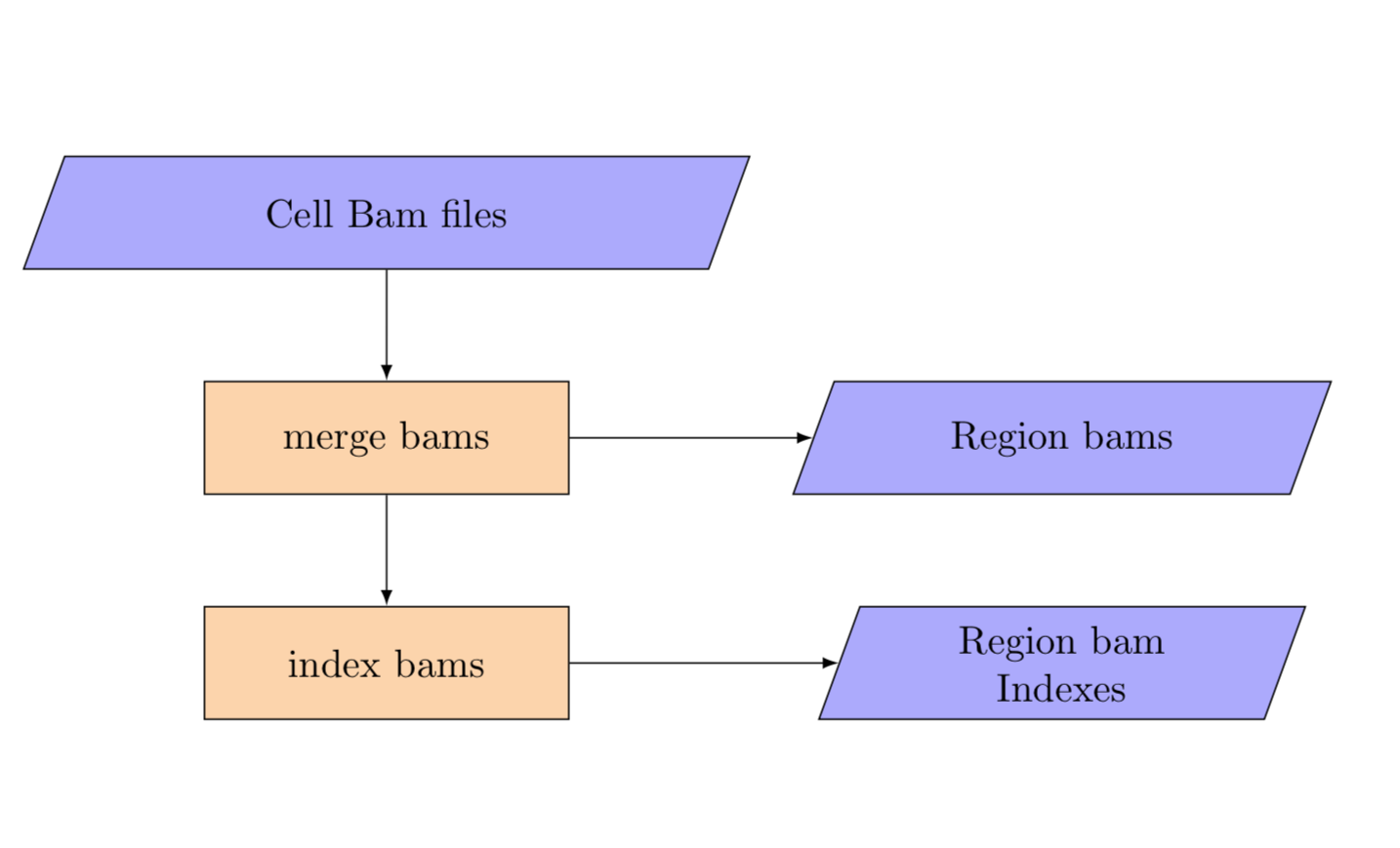 merge_cell_bams
merge_cell_bams
The tumour needs to be simultaneously merged across cells and split by region. The input for this step is the per cell bam yaml and the template for the merged bams by region.
Input:¶
cell_bams:
SA123X5-A12345-R04-C03:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C03.bam
SA123X5-A12345-R04-C05:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C05.bam
SA123X5-A12345-R04-C07:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C07.bam
SA123X5-A12345-R04-C09:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C09.bam
SA123X5-A12345-R04-C10:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C10.bam
Run:¶
single_cell merge_cell_bams \
--input_yaml inputs/SC-1234/merge_cell_bams.yaml \
--tmpdir temp/SC-1234/tmp \
--pipelinedir pipeline/SC-1234 \
--out_dir results/SC-1234/results \
...
Outputs¶
The split merge pipeline produces a set of bams split by region.
Region BAMs¶
Bam files are named as {REGION_1}.bam in the BAM results directory.
The metadata file is structured as follows:
filenames:
- {REGION_1}.bam
- {REGION_2}.bam
...
meta:
command: 'single_cell merge_cell_bams ...'
input_yaml: input.yaml
type: pseudowgs_regionbams
version: v0.0.0
cell_ids:
- CELL_ID_1
- CELL_ID_1
bams:
template: {region}.bam
instances:
- region: REGION_1
- region: REGION_2
5. Split bams¶
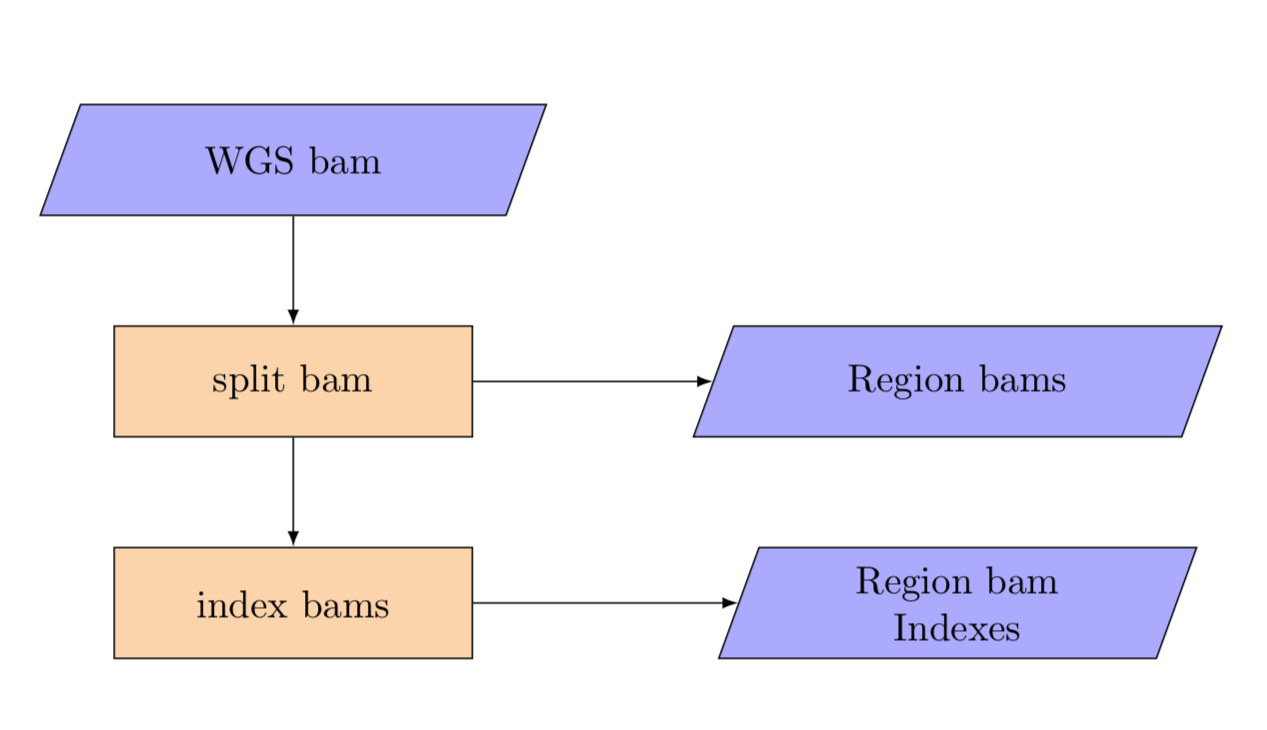 split_wgs_bam
split_wgs_bam
The normal also needs to be split by region from an input data path to an output per region template.
Input:¶
normal:
bam: scdnadev/testdata/pseudobulk/DAH370N_filtered.bam
Run:¶
single_cell split_wgs_bam \
--input_yaml inputs/SC-1234/merge_cell_bams.yaml \
--tmpdir temp/SC-1234/tmp \
--pipelinedir pipeline/SC-1234 \
--out_dir results/SC-1234/results \
...
Outputs¶
The split pipeline produces a set of bams split by region.
Region BAMs¶
Bam files are named as {REGION_1}.bam in the BAM results directory.
The metadata file is structured as follows:
filenames:
- {REGION_1}.bam
- {REGION_2}.bam
...
meta:
command: 'single_cell split_wgs_bam ...'
input_yaml: input.yaml
type: wgs_regionbams
version: v0.0.0
bams:
template: {region}.bam
instances:
- region: REGION_1
- region: REGION_2
6. Variant Calling¶
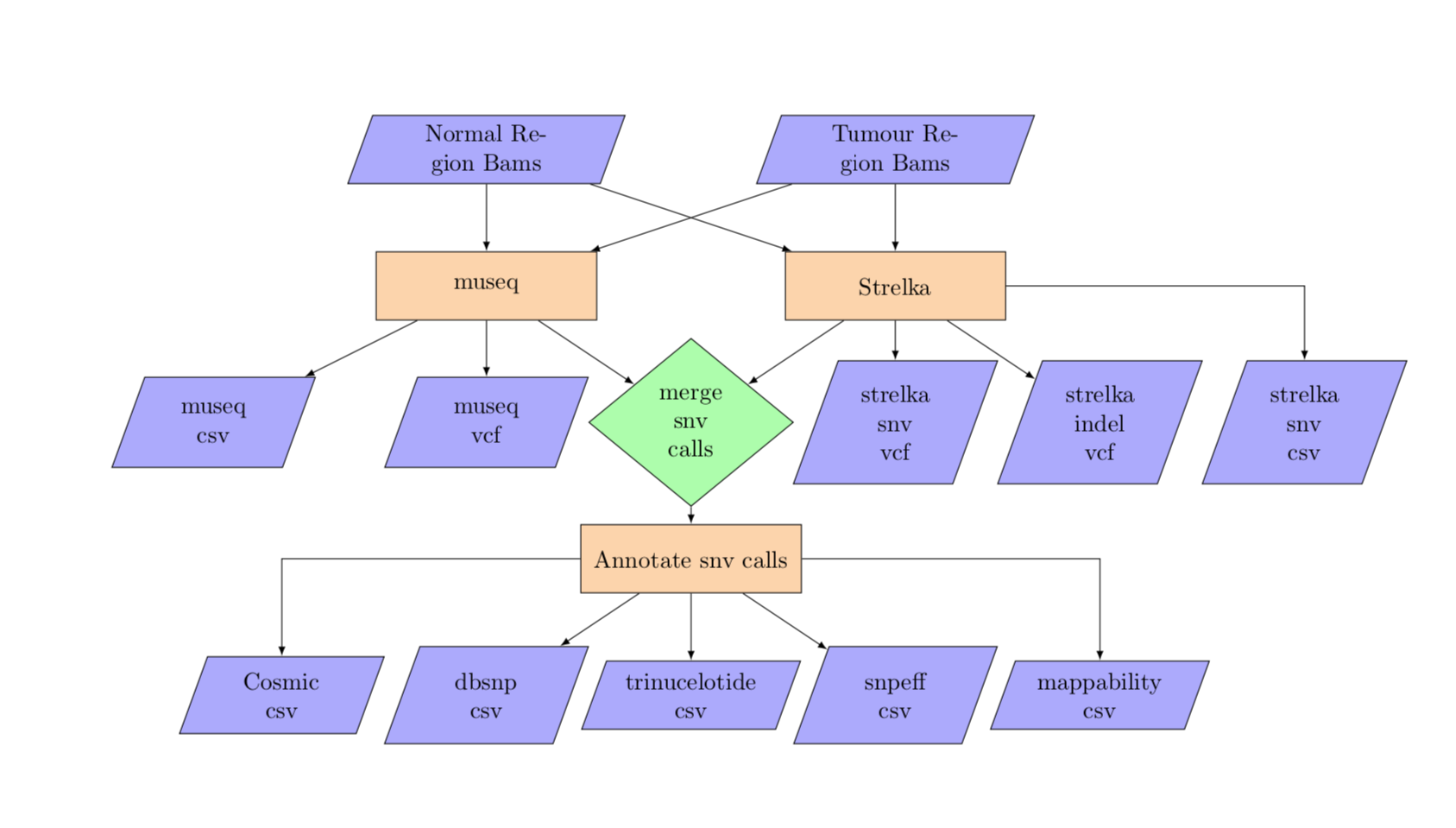 variant_calling
variant_calling
Inputs are a WGS tumour bam file and a WGS normal bam file along with the tumour cells. The bam files are used for the variant calling. The pipeline also generates counts at the snvs for each cell. The variant calling takes in both the per cell bam yaml, using the per cell bams for variant allele counting, and the tumour and normal region templates for calling snvs in parallel by region.
Input:¶
normal:
1-1-10000000:
bam: split_wgs_bam/1-1-10000000_split_wgs.bam
1-100000001-110000000:
bam: split_wgs_bam/1-100000001-110000000_split_wgs.bam
1-10000001-20000000:
bam: split_wgs_bam/1-10000001-20000000_split_wgs.bam
...
tumour:
1-1-10000000:
bam: merge_cell_bams/1-1-10000000_split_wgs.bam
1-100000001-110000000:
bam: merge_cell_bams/1-100000001-110000000_split_wgs.bam
1-10000001-20000000:
bam: merge_cell_bams/1-10000001-20000000_split_wgs.bam
...
Run:¶
single_cell variant_calling \
--input_yaml inputs/SC-1234/variant_calling.yaml \
--tmpdir temp/SC-1234/tmp \
--pipelinedir pipeline/SC-1234 \
--out_dir results/SC-1234/results \
...
Outputs¶
The metadata file is structured as follows:
filenames:
- {LIBRARY_ID}_{TABLE_1}.csv.gz
- {LIBRARY_ID}_{TABLE_1}.csv.gz.yaml
...
meta:
command: 'single_cell variant_calling ...'
input_yaml: input.yaml
type: variant_calling
version: v0.0.0
7. Breakpoint calling¶
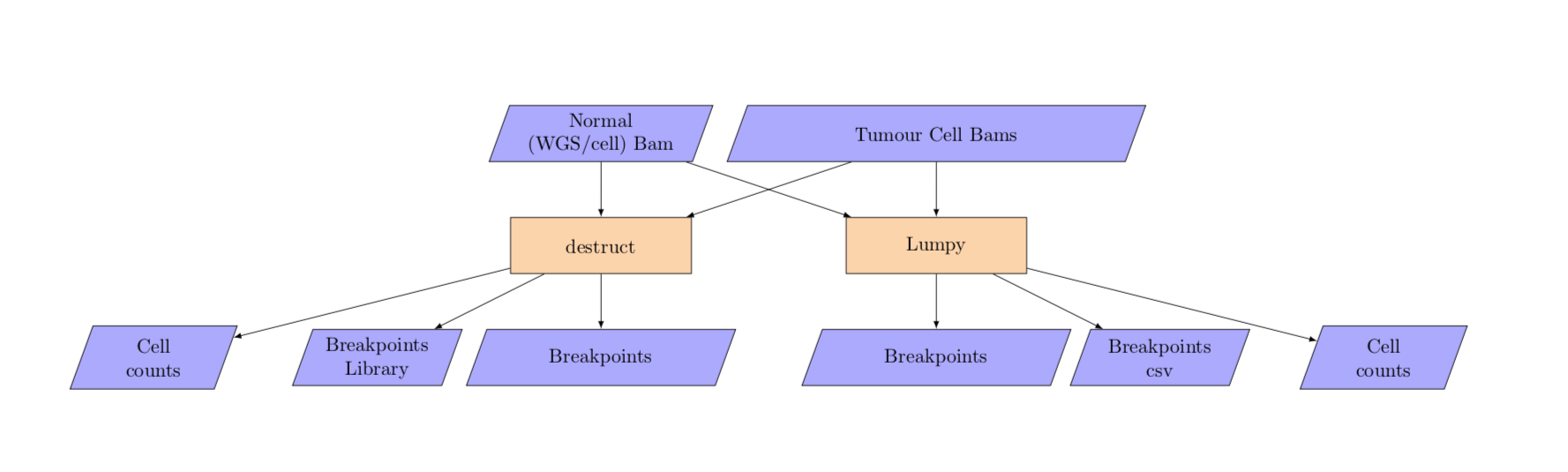 breakpoint_calling
breakpoint_calling
The breakpoint analysis takes in per cell bam yaml in addition to the unsplit matched normal bam filename.
Input:¶
yaml file format with a single normal bam¶
normal_wgs:
bam: singlecelldatatest/testdata/pseudobulk/SA123N.bam
tumour_cells:
SA123X5-A12345-R04-C03:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C03.bam
SA123X5-A12345-R04-C05:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C05.bam
SA123X5-A12345-R04-C07:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C07.bam
SA123X5-A12345-R04-C09:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C09.bam
SA123X5-A12345-R04-C10:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C10.bam
yaml file format with a cells for normal¶
normal_cells:
SA123N-A12345-R04-C03:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C03.bam
SA123N-A12345-R04-C05:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C05.bam
SA123N-A12345-R04-C07:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C07.bam
SA123N-A12345-R04-C09:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C09.bam
SA123N-A12345-R04-C10:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C10.bam
tumour_cells:
SA123X5-A12345-R04-C03:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C03.bam
SA123X5-A12345-R04-C05:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C05.bam
SA123X5-A12345-R04-C07:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C07.bam
SA123X5-A12345-R04-C09:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C09.bam
SA123X5-A12345-R04-C10:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C10.bam
Run:¶
single_cell breakpoint_calling \
--input_yaml inputs/SC-1234/breakpoint_calling.yaml \
--tmpdir temp/SC-1234/tmp \
--pipelinedir pipeline/SC-1234 \
--out_dir results/SC-1234/results \
...
NOTE: The input bam files for lumpy must be aligned with bwa mem.
Outputs¶
The metadata file is structured as follows:
filenames:
- {LIBRARY_ID}_{TABLE_1}.csv.gz
- {LIBRARY_ID}_{TABLE_1}.csv.gz.yaml
...
meta:
command: 'single_cell breakpoint_calling ...'
input_yaml: input.yaml
type: breakpoint_calling
version: v0.0.0
8. Haplotype calling:¶
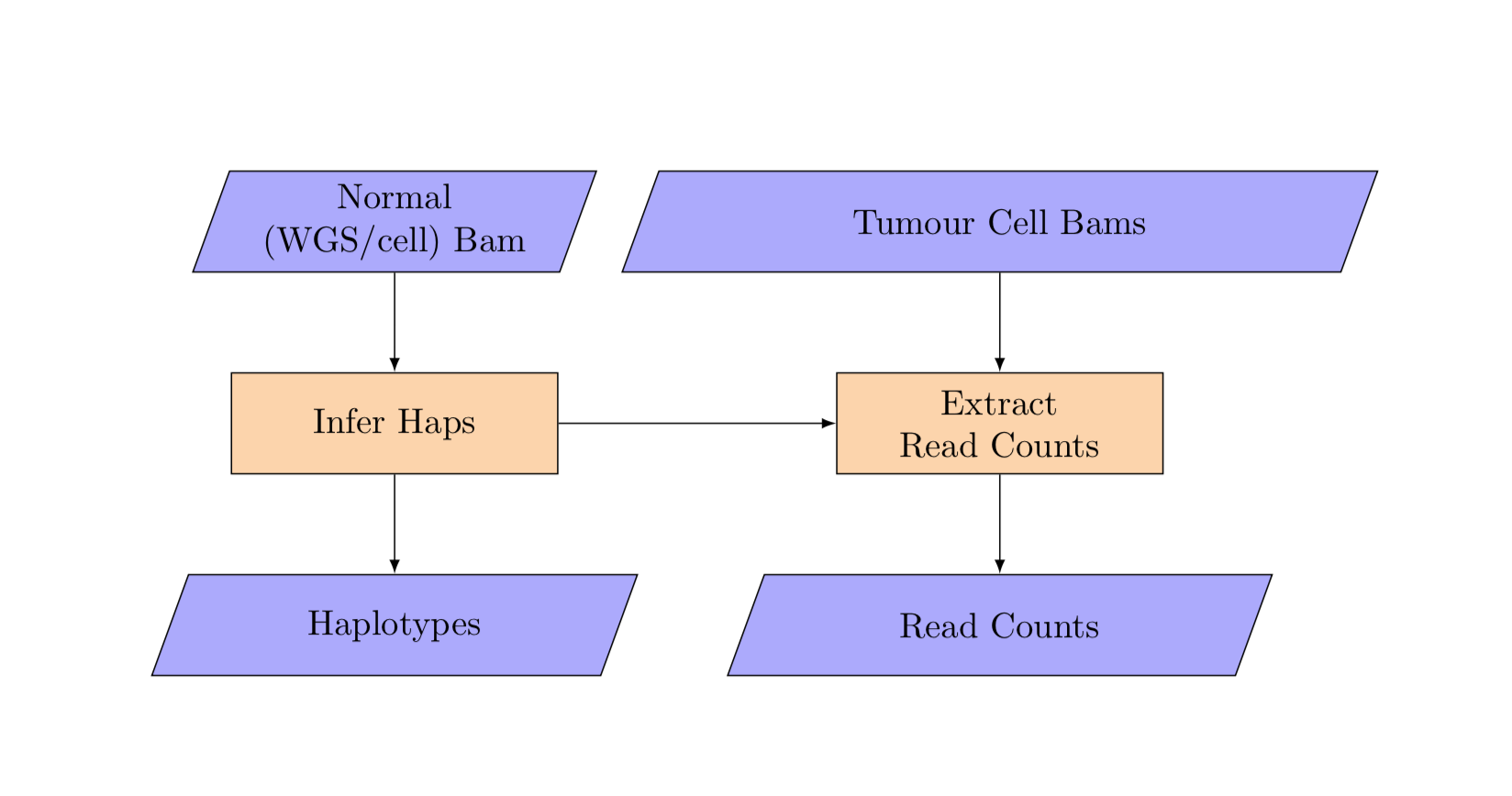 infer_haps
infer_haps
The haplotype analysis takes in per cell bam yaml in addition to the unsplit matched normal bam filename.
Input:¶
yaml file format with a single normal bam¶
normal_wgs:
bam: singlecelldatatest/testdata/pseudobulk/SA123N.bam
tumour_cells:
SA123X5-A12345-R04-C03:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C03.bam
SA123X5-A12345-R04-C05:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C05.bam
SA123X5-A12345-R04-C07:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C07.bam
SA123X5-A12345-R04-C09:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C09.bam
SA123X5-A12345-R04-C10:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C10.bam
yaml file format with a cells for normal¶
normal_cells:
SA123N-A12345-R04-C03:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C03.bam
SA123N-A12345-R04-C05:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C05.bam
SA123N-A12345-R04-C07:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C07.bam
SA123N-A12345-R04-C09:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C09.bam
SA123N-A12345-R04-C10:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C10.bam
tumour_cells:
SA123X5-A12345-R04-C03:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C03.bam
SA123X5-A12345-R04-C05:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C05.bam
SA123X5-A12345-R04-C07:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C07.bam
SA123X5-A12345-R04-C09:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C09.bam
SA123X5-A12345-R04-C10:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C10.bam
Run:¶
single_cell haplotype_calling \
--input_yaml inputs/SC-1234/haplotype_calling.yaml \
--tmpdir temp/SC-1234/tmp \
--pipelinedir pipeline/SC-1234 \
--out_dir results/SC-1234/results \
...
Outputs¶
The metadata file is structured as follows:
filenames:
- {LIBRARY_ID}_{TABLE_1}.csv.gz
- {LIBRARY_ID}_{TABLE_1}.csv.gz.yaml
...
meta:
command: 'single_cell infer_haps ...'
input_yaml: input.yaml
type: infer_haps
version: v0.0.0
9. SNV genotyping:¶
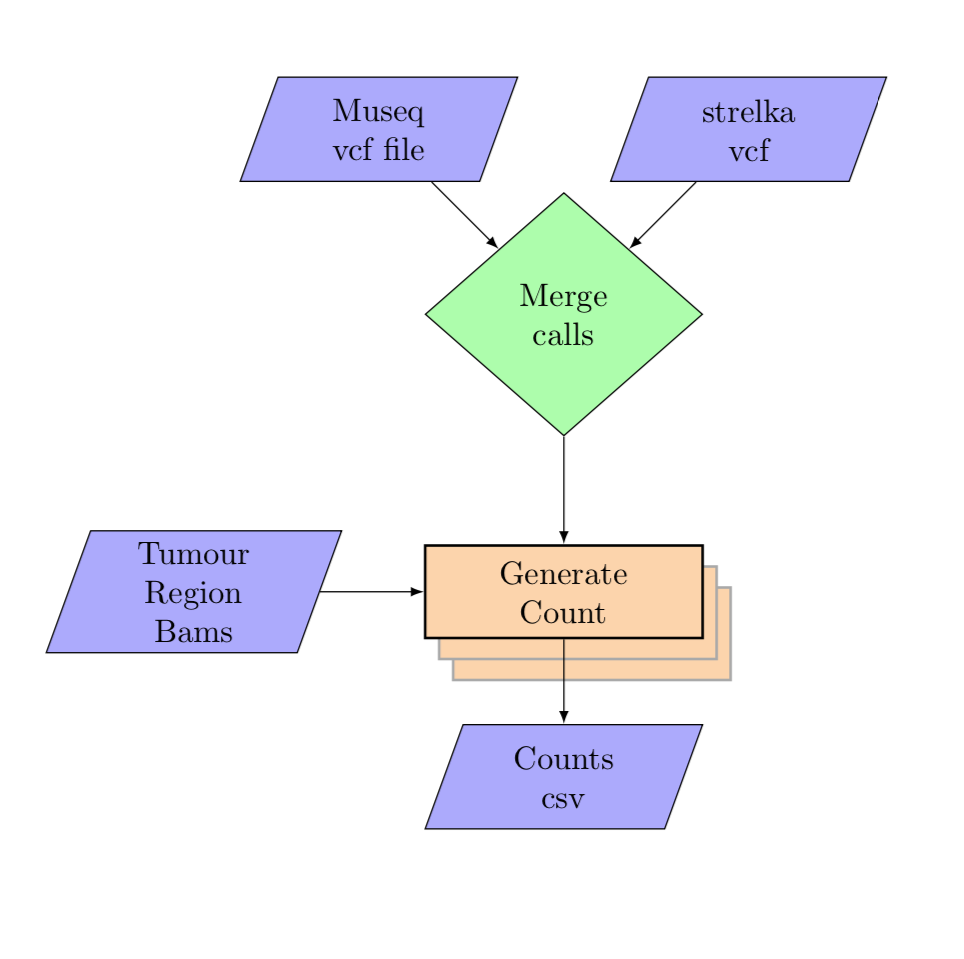 variant_counting
variant_counting
Input:¶
vcf_files:
SA123N:
A1234:
museq_vcf: vcfs/museq.vcf
strelka_snv_vcf: vcfs/strelka.vcf
tumour_cells:
SA123N:
SA123N-A12345-R04-C03:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C03.bam
SA123N-A12345-R04-C05:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C05.bam
SA123N-A12345-R04-C07:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C07.bam
SA123N-A12345-R04-C09:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C09.bam
SA123N-A12345-R04-C10:
bam: data/single_cell_indexing/bam/A12345/grch37/bwa-aln/SA123X5-A12345-R04-C10.bam
Run:¶
single_cell snv_genotyping \
--input_yaml inputs/SC-1234/snv_genotyping.yaml \
--tmpdir temp/SC-1234/tmp \
--pipelinedir pipeline/SC-1234 \
--out_dir results/SC-1234/results \
...
Outputs¶
The metadata file is structured as follows:
filenames:
- {LIBRARY_ID}_{TABLE_1}.csv.gz
- {LIBRARY_ID}_{TABLE_1}.csv.gz.yaml
...
meta:
command: 'single_cell snv_genotyping ...'
input_yaml: input.yaml
type: snv_genotyping
version: v0.0.0
10. Germline Calling:¶
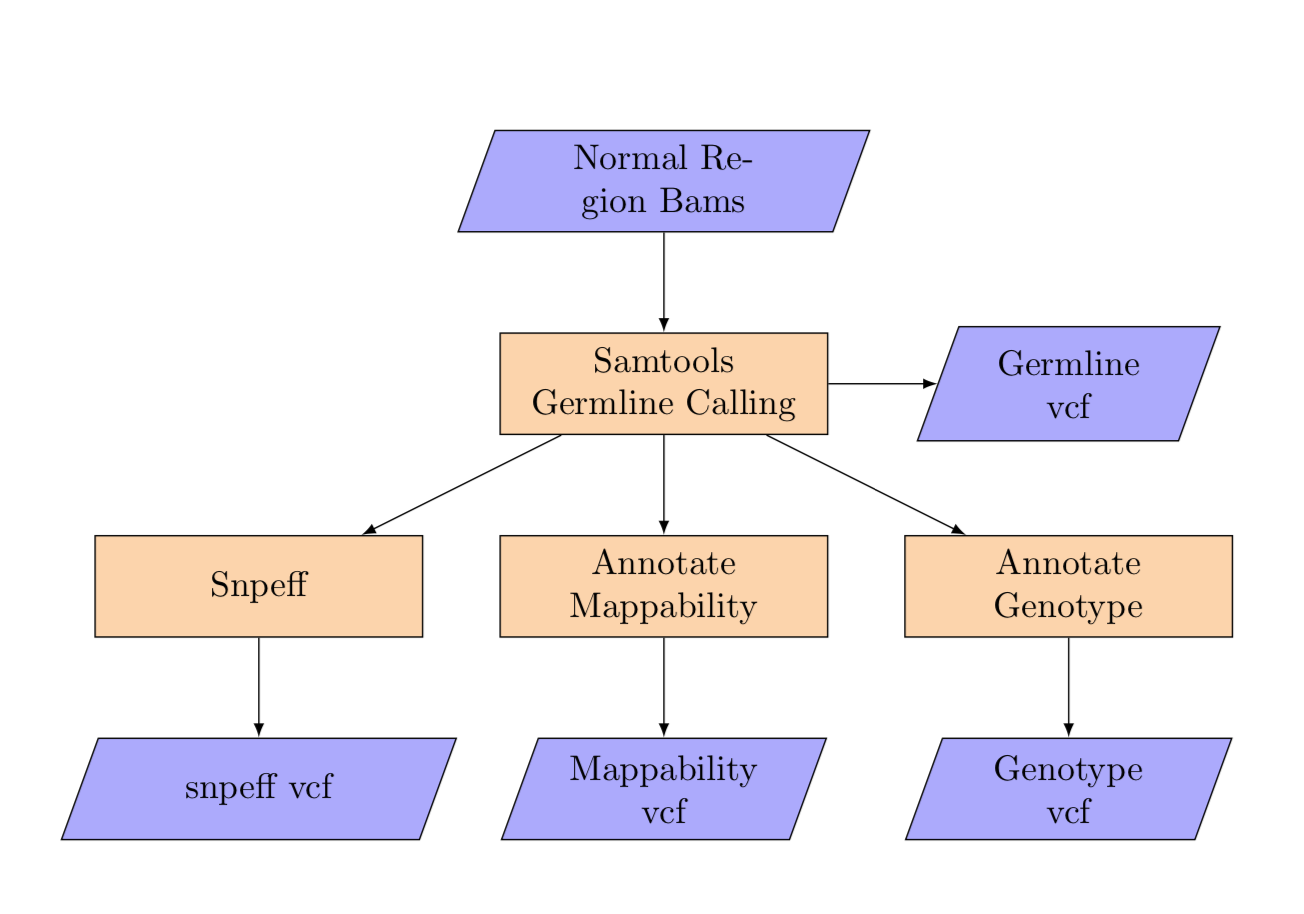 germline_calling
germline_calling
Input:¶
normal:
1-1-10000000:
bam: split_wgs_bam/1-1-10000000_split_wgs.bam
1-100000001-110000000:
bam: split_wgs_bam/1-100000001-110000000_split_wgs.bam
1-10000001-20000000:
bam: split_wgs_bam/1-10000001-20000000_split_wgs.bam
...
Run:¶
single_cell germline \
--input_yaml inputs/SC-1234/variant_counting.yaml \
--tmpdir temp/SC-1234/tmp \
--pipelinedir pipeline/SC-1234 \
--out_dir results/SC-1234/results \
...
Outputs¶
The metadata file is structured as follows:
filenames:
- {LIBRARY_ID}_{TABLE_1}.csv.gz
- {LIBRARY_ID}_{TABLE_1}.csv.gz.yaml
...
meta:
command: 'single_cell germline_calling ...'
input_yaml: input.yaml
type: germline_calling
version: v0.0.0
11. Pseudobulk QC:¶
Input:¶
SA123:
SA123X1:
A123:
mappability: PSEUDO_BULK_QC/ref_test_data/snv_mappability.csv.gz
strelka: PSEUDO_BULK_QC/ref_test_data/snv_strelka.csv.gz
museq: PSEUDO_BULK_QC/ref_test_data/snv_museq.csv.gz
cosmic_status: PSEUDO_BULK_QC/ref_test_data/snv_cosmic_status.csv.gz
snpeff: PSEUDO_BULK_QC/ref_test_data/snv_snpeff.csv.gz
dbsnp_status: PSEUDO_BULK_QC/ref_test_data/snv_dbsnp_status.csv.gz
trinuc: PSEUDO_BULK_QC/ref_test_data/snv_trinuc.csv.gz
counts: PSEUDO_BULK_QC/ref_test_data/snv_genotyping_counts.csv.gz
destruct_breakpoint_annotation: PSEUDO_BULK_QC/ref_test_data/destruct_breakpoints.csv.gz
destruct_breakpoint_counts: PSEUDO_BULK_QC/ref_test_data/destruct_cell_counts.csv.gz
lumpy_breakpoint_annotation: PSEUDO_BULK_QC/ref_test_data/lumpy_breakpoints.csv.gz
lumpy_breakpoint_evidence: PSEUDO_BULK_QC/ref_test_data/lumpy_breakpoints_evidence.csv.gz
haplotype_allele_data: PSEUDO_BULK_QC/ref_test_data/allele_counts.csv.gz
annotation_metrics: PSEUDO_BULK_QC/ref_test_data/annotation_metrics.csv.gz
hmmcopy_reads: PSEUDO_BULK_QC/ref_test_data/hmmcopy_reads.csv.gz
hmmcopy_segs: PSEUDO_BULK_QC/ref_test_data/hmmcopy_segments.csv.gz
hmmcopy_metrics: PSEUDO_BULK_QC/ref_test_data/hmmcopy_metrics.csv.gz
alignment_metrics: PSEUDO_BULK_QC/ref_test_data/alignment_metrics.csv.gz
gc_metrics: PSEUDO_BULK_QC/ref_test_data/gc_metrics.csv.gz
indel_file: PSEUDO_BULK_QC/ref_test_data/strelka_indel.vcf.gz
Run:¶
single_cell pseudo_bulk_qc \
--input_yaml inputs/SC-1234/variant_counting.yaml \
--tmpdir temp/SC-1234/tmp \
--pipelinedir pipeline/SC-1234 \
--out_dir results/SC-1234/results \
...
Outputs¶
The metadata file is structured as follows:
filenames:
./{patient_id}/{sample_id}/{library_id}_snvs_high_impact.csv
./{patient_id}/{sample_id}/{library_id}_num_cells_class_top10_mutsig.pdf
./{patient_id}/{sample_id}/{library_id}_num_cells_class_mutsig.pdf
./{patient_id}/{sample_id}/{library_id}/snvs_high_impact.csv
./{patient_id}/{sample_id}/{library_id}/samplelevelmaf.maf
./{patient_id}/{sample_id}/{library_id}/snv_alt_counts.pdf
./{patient_id}/{sample_id}/{library_id}/snvs_all.csv
./{patient_id}/{sample_id}/{library_id}/summary.csv
./{patient_id}/{sample_id}/{library_id}/datatype_summary.csv
./{patient_id}/{sample_id}/{library_id}/snv_cell_counts.pdf
./{patient_id}/{sample_id}/{library_id}/trinuc.csv
./{patient_id}/{sample_id}/{library_id}_snv_genome_count.pdf
./{patient_id}/{sample_id}/{library_id}_adjacent_distance_class_mutsig.pdf
./{patient_id}/{sample_id}/{library_id}_adjacent_distance_class_top10_mutsig.pdf
./{patient_id}/{sample_id}/{library_id}_snv_adjacent_distance.pdf
meta:
command: 'single_cell pseudo_bulk_qc ...'
input_yaml: input.yaml
type: pseudo_bulk_qc
version: v0.0.0
11. Generate Config¶
The pipeline auto generates a config file with the default parameters before every run. Some of the values in the config file can be updated by using the --config_override option. generate_config option allows users to generate the config files. These configs can then be specified as input to the pipeline after making the required changes.
single_cell generate_config --pipeline_config pipeline_config.yaml --batch_config batch_config.yaml
the pipeline config file contains all pipeline defaults and the batch config specifies all azure batch specific settings.
the pipeline config can be specified manually when running the pipeline with --config_file option and the batch config with --submit_config option.
12. Clean Sentinels¶
the pipeline will skip any successful tasks from previous runs when run again. The --rerun flag force run all tasks including the successful tasks from the previous runs while the clean_sentinels option provides a more fine grained control.
single_cell clean_sentinels --mode list --pattern "*" --tmpdir temp/SC-1234/tmp/hmmcopy
will list all successfully completed hmmcopy tasks
single_cell clean_sentinels --mode delete --pattern "*" --tmpdir temp/SC-1234/tmp/hmmcopy
is the same as --rerun flag
running
single_cell clean_sentinels --mode delete --pattern "*plot_heatmap*" --tmpdir temp/SC-1234/tmp/hmmcopy
before launching the hmmcopy will rerun the heatmap plotting and any tasks that haven’t completed yet.
Common options¶
- add
--storage azureblobto read the inputs and write the outputs to Microsoft Azure Blob Storage - add
--submit localto run the pipeline on the current node - add
--submit asyncqsubto run the pipeline on a SGE cluster - add
--submit azurebatchto run the tasks on Microsoft Azure Batch - add
--nocleanupto disable temporary file deletion - add
--loglevel DEBUGfor verbose logging --submit_config <path to yaml config>for custom azure batch submission configuration file. see 10. Generate config for more details--config_file <path to yaml config>to specify pipeline config file.